

Электровыделение металлов |
Механизмы электролитической диссоциации в неводных растворах
Механизм электролитической диссоциации, т. е. природа ионов, образующихся в системе электролит—растворитель и участвующих в переносе тока, стал предметом исследований лишь в последнее десятилетие. Причины несколько запоздалого обращения к столь важной проблеме заключаются, по-видимому, в том, что природа ионов, образующихся при электролитической диссоциации в водных растворах, часто представлялась априорно очевидной, и эта очевидность механически переносилась на неводные растворы. Углубленное изучение схемы возникновения электролитного раствора, в частности термодинамические исследования, показало, что даже в водных растворах установление чисел гидратации, границ ближней и дальней сольватации имеет решающее значение для полного описания электролитного раствора. В неводных же средах, где, в отличие от большинства водных растворов, в системе электролит — растворитель присутствует намного больше равновесных форм (см. схему (1—14)), определение природы и состава ионов имеет первостепенное значение для понимания процессов, происходящих в системе. Очевидно также и прикладное значение проблемы природы ионов в неводных растворах: вряд ли процесс электроосаждения металлов из неводных растворов можно эффективно осуществлять, если не известна эта важнейшая характеристика системы.
Для исследований использовали такие эффективные методы изучений неводных растворов, как ЯМР-спектроскопию на ядрах атомов, входящих в состав молекулы растворителя L-кислоты, ИК-спектроскопию, рентгеновскую дифракто-скопию, метод меченых атомов.
Схемы электролитической диссоциации, приводимые в литературе, могут быть подразделены на четыре основных типа:
1. Ионизация I. Kислоты. Все L-кислоты проявляют достаточно четкую способность к автоионизации:

Сольватация (основным растворителем) смещает равновесие диссоциации L-кислоты вправо:

В тех случаях, когда кислота находится в избытке, анион сольватируется молекулой кислоты

Отсутствие (либо весьма малая степень) сольватации аниона в схеме (1—69, а) справедливо в случае наиболее часто применяющихся в практике основных растворителей; если же процесс диссоциации протекает по схеме (1—69, б), то отсутствие сольватации аниона объясняется тем, что его центральный атом координационно насыщен.
Рассматриваемая схема приводится во многих работах М. И. Усановича и его школы. Так, возникновение ионов в растворах трихлорида сурьмы в абсолютной уксусной кислоте описывается уравнениями:

Аналогичный механизм диссоциации предлагается и для растворов L-кислот в спиртах и сложных эфирах. С последней работой согласуется и точка зрения С. П. Мискиджьяна.
О. А. Осипов предложил схему (1—69, б) для определения природы ионов в системе пентахлорид пиобия — сложные эфиры. В работах на основе данной схемы объяснено возникновение ионов в растворах галогенидов алюминия в разнообразных органических растворителях. Аналогичные схемы можно найти в литературе в приложении к галогенидам многих иных металлов.


В работах Ю. А. Лысенко отстаивается следующий вариант схемы (1—69 б):


Отметим малую вероятность реализации подобной схемы, так как она предусматривает, что электронодонорный растворитель в одинаковой степени сольватирует и электронодефицитный катион и анион-основание, причем последний к тому же координационно насыщен анионами галоида.
2. Ионизация растворителя. Способность жидкости подвергаться процессу автоионизации:

 лиата) в индивидуальной жидкости связана с распределением при данной температуре молекул по энергиям, с макродиэлектрической проницаемостью жидкости и другими факторами.
лиата) в индивидуальной жидкости связана с распределением при данной температуре молекул по энергиям, с макродиэлектрической проницаемостью жидкости и другими факторами.
В зависимости от природы растворителя процесс автоионизации передается одной из следующих схем:

 некоторых растворителей.
некоторых растворителей.

Введение L-кислоты в растворитель обычно сдвигает вправо равновесие процесса автоионизации (1—70):

Для часто встречающегося случая растворителей с протолитическим равновесием процесс (1—72) выразится уравнением


 в ароматических карбо-
в ароматических карбо-
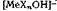 и RCO+.
и RCO+.
Сольволиз. Под сольоволизом в данном случае понимается процесс, приводящий к образованию неэлектролитных продуктов. С учетом обозначений, введенных в схеме (1—70), процесс можно представить следующим образом:

Протекание сольволиза в системе не означает, что не образуются электролитные соединения, но возникновение последних является уже следствием последующих вторичных процессов:


Сольволиз относится к числу наиболее распространенных процессов в системах с участием L-кислот. Ниже приводится несколько примеров сольволиза галогенидов металлов различными растворителями.
 . Примером сольволиза в гидразине может служить реакция
. Примером сольволиза в гидразине может служить реакция


 В жидком фтористом водороде сольволиз различных солей металлов чаще всего приводит к образованию соответствующих фторидов.
В жидком фтористом водороде сольволиз различных солей металлов чаще всего приводит к образованию соответствующих фторидов.
 . Наиболее обстоятельно процессы сольволиза изучены в спиртах и карбоновых кислотах.
. Наиболее обстоятельно процессы сольволиза изучены в спиртах и карбоновых кислотах.
 установлено образование двух продуктов сольволиза — моно- и дибутоксизамещенных.
установлено образование двух продуктов сольволиза — моно- и дибутоксизамещенных.
Сольволиз в системах трихлорид сурьмы — алифатические спирты достаточно подробно описан в работах, аналогичный процесс в системах, образованных пеитахлоридом сурьмы с этанолом либо уксусной кислотой, изучен в работе. Сольволизные процессы трихлорида железа в спиртах рассмотрены в работе.
 .
.
Сольволиз в карбоновых кислотах также был предметом многих исследований. Изучался сольволиз хлорного олова, трихлорида и пентахлорида сурьмы, трихлорида железа. Подробное исследование продуктов сольволиза солей алюминия, титана, циркония, тория, олова, сурьмы и теллура в монохлоруксусной кислоте предпринято в работе.
Образование карбкатионо в. К реакциям этого типа относятся процессы, протекающие по схеме

 авторы работ схемой
авторы работ схемой
 с галоидангид-
с галоидангид-
ридами карбоновых кислот.
Аналогичный механизм взаимодействия в системах трихлорид сурьмы— алифатические спирты, предположенный одним из авторов совместно с В. П. Басовым в работах, по-видимому, не верен, так как трактовка результатов эксперимента ПМР не позволяет сделать однозначный выбор между обсуждаемой схемой и схемами (1—73) и (1—74).
 ни с анионом.
ни с анионом.
Обстоятельное сопоставление различных схем диссоциации галогенидов Sn (IV) и Sb (V) приведено в обзоре.
 _ (очевидным условием протекания такого рода взаимодействия является отсутствие растворителя, обычно нивелирующего силу L-кис лот).
_ (очевидным условием протекания такого рода взаимодействия является отсутствие растворителя, обычно нивелирующего силу L-кис лот).
В системах указанного типа обнаружен помимо приведенного еще весьма своеобразный механизм взаимодействия, названный ион-радикальным:
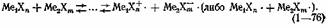
Возникновение радикалов в системах, образованных галогенидами алюминия либо галлия с полухлористой (либо полубромистой) серой, было установлено с помощью ЭПР-спектроскопии, причем было найдено, что максимум интенсивности сигнала ЭПР приходится на состав раствора, отвечающий стехиометрии взаимодействия.
Существующие методы исследования природы взаимодействия в системах L-кислоты — основный органический растворитель не всегда позволяют выбрать какую-либо определенную схему диссоциации. Поэтому при выборе наиболее достоверной схемы необходимо нередко прибегать к анализу химических особенностей системы. При этом существенную роль играет природа основания, а именно его способность принимать участие в кислотно-основном взаимодействии только в молекулярной форме (апротонные основания) либо также или даже исключительно — в ионной форме (протолитические основания).
Исследование различных систем, образованных Н- и L-кислота-ми, с одной стороны, и апротонными либо протолитическими основаниями— с другой, позволило выявить некоторые общие закономерности, имеющие значение для выбора наиболее достоверной схемы взаимодействия.
 , ЯМР- и ИК-спектро-скопией.
, ЯМР- и ИК-спектро-скопией.

Таким образом, наиболее общая схема взаимодействия в системах L-кислота — протолитическое основание должна передаваться схемами — для области, богатой растворителем:

 Справедливость этой схемы доказана экспериментами по электропереносу радиоизотопов: в то время как радиохлор мигрирует только к аноду, радиоуглерод и тритий — только к катоду; в областях, богатых L-кислотой, к аноду мигрирует также радиожелезо.
Справедливость этой схемы доказана экспериментами по электропереносу радиоизотопов: в то время как радиохлор мигрирует только к аноду, радиоуглерод и тритий — только к катоду; в областях, богатых L-кислотой, к аноду мигрирует также радиожелезо.
1.5.4. Механизмы переноса тока в неводных растворах
В неводиых растворах перенос тока может реализовываться за счет каждого из трех основных механизмов переноса тока в растворах— ионмиграционного, ионотропного и электронного.
Ионотропный механизм переноса тока, известный для водных растворов кислот (прототропный механизм, эстафетный механизм), энергетически более выгоден, чем миграция гидратированного протона.
Исследования последних лет показали, что прототропный механизм переноса тока в протолитических системах встречается гораздо чаще, чем это предполагали. Значительная доля тока ( по ряду признаков иногда достигающая 1) переносится по прото-тропному механизму через жидкие индивидуальные сильные Н-кислоты — серную, селеновую, ортофосфорную. Соответственно по прототропному механизму переносится ток в водных и неводных растворах этих кислот (в областях концентраций с преобладанием кислотного компонента). Имеются достаточно веские аргументы в пользу того, что в некоторых случаях ток в карбоновых кислотах как растворителях может также переноситься по прототропному механизму. После воды наиболее подробно прототропный механизм переноса тока изучен для растворов в алифатических спиртах.
С возможностью реализации прототропного (наряду с ионмиг-рационным) механизма переноса тока всегда необходимо считаться, когда идет речь о растворах L-кислот в протолитических растворителях, в которых в основном протекают процессы сольволиза, приводящие к возникновению в растворе сольватированного протона (см. схему (1—77)).
В отличие от водных в неводных растворах ионотропный механизм переноса тока возможен и для более тяжелых, чем протон, ионов.
 и об йодотропном механизме проводимости в растворах йодидов щелочных металлов в жидком йоде.
и об йодотропном механизме проводимости в растворах йодидов щелочных металлов в жидком йоде.
 0,8—0,9 мол. доли.
0,8—0,9 мол. доли.
Для разработки оптимального режима электролиза немаловажное значение имеет определение соотносительного вклада ион-миграционного и ионотропного механизмов переноса тока.
 .
.
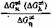 в
в
качестве критерия вклада прототропного механизма проводимости: при полностью ионмиграционном механизме это отношение равно единице; при сосуществовании ионмиграционного и прототропного механизмов переноса тока это отношение менее единицы, причем, чем выше вклад прототропного механизма, тем в большей степени отношение энергий активации отклоняется от единицы.
Указанное отношение может служить критерием соотносительного вклада ионмиграционного и галогенотропного механизмов переноса тока, что можно иллюстрировать рядом систем типа три-морид сурьмы—апротонные основания, в которых наличие галогенотропного механизма переноса тока было доказано прямым экспериментом.
 составляет приблизительно 0,9.
составляет приблизительно 0,9.
Для разбавленных водных растворов достаточно определенным
 приближающееся к двум в растворах с доминирующим прототропным механизмом переноса тока.
приближающееся к двум в растворах с доминирующим прототропным механизмом переноса тока.
 . При сосуществовании обоих механизмов эта сумма меньше единицы, причем, тем значительнее, чем выше вклад ионотропного механизма переноса тока. Следует, впрочем, заметить, что экспериментальное определение чисел переноса в неводных растворах с удовлетворительной точностью — задача непростая.
. При сосуществовании обоих механизмов эта сумма меньше единицы, причем, тем значительнее, чем выше вклад ионотропного механизма переноса тока. Следует, впрочем, заметить, что экспериментальное определение чисел переноса в неводных растворах с удовлетворительной точностью — задача непростая.
![]()


где значение а для каждой системы определяется прямым, например ИК-спектроскопическим, экспериментом.
Электронный механизм проводимости, встречающийся в растворах щелочных металлов в сильноосновных растворителях, где время жизни сольватированного электрона (сильнейшее основание) достаточно велико, к предмету этой книги сколько нибудь прямого отношения не имеет. Поэтому ограничимся лишь упоминанием об этом механизме переноса тока.

